Medical Device Warehouse Requirements: The Complete FDA & ISO Compliance Guide (2026)

Managing a medical device warehouse is far more complex than running a standard distribution center. With patient safety, regulatory scrutiny, and supply chain integrity all on the line, operators and brand owners must navigate a dense web of federal regulations, international standards, and quality management requirements before a single unit ships.
Whether you are a device manufacturer evaluating storage facilities, a logistics director building out a new fulfillment network, or a startup preparing for market launch, understanding medical device warehouse requirements is non-negotiable. This guide covers everything, from FDA registration and 21 CFR Part 820 compliance to ISO 13485 certification, temperature control, traceability, and how to select the right order fulfillment solutions partner for regulated healthcare products.
Quick Stat: The global medical device warehouse and logistics market was valued at $43.03 billion in 2024 and is projected to grow at a CAGR of 4.70% through 2032, driven by rising regulatory complexity and increasing demand for specialized storage solutions.
What Makes Medical Device Warehousing Different?
Not all warehouses are created equal. A facility storing consumer electronics has almost nothing in common with one storing Class III implantable devices. Medical device warehousing carries unique obligations because failures whether from contamination, improper temperature handling, or traceability breakdowns, can directly harm patients.
The core differences from general warehousing include:
• Regulatory oversight: FDA, ISO, OSHA, IATA, and state-level health authorities all have jurisdiction depending on device type.
• Traceability requirements: Every unit must be trackable from receipt through delivery and, in some cases, post-market surveillance.
• Environmental controls: Temperature, humidity, and air quality must be monitored, validated, and documented continuously.
• Quality Management Systems (QMS): Warehouses operating under a device manufacturer’s establishment registration must maintain documented quality systems.
• Chain-of-custody documentation: All receipt, storage, and dispatch activities must be recorded to support regulatory inspections and product recalls.
Because of this complexity, many manufacturers partner with specialized 3PL fulfillment providers that are already built to handle FDA-regulated product categories.
FDA Registration Requirements for Medical Device Warehouses
FDA registration is the foundational requirement for any facility storing medical devices in the United States. Under 21 CFR Part 807, device establishments, including warehouses and distribution facilities, must register with the FDA and list the devices they handle annually.
Who Must Register?
• Manufacturers of finished medical devices
• Initial importers of foreign-manufactured devices
• Contract manufacturers and sterilizers
• Repackagers and relabelers
• Third-party warehouses operating under a manufacturer’s QMS
Registration must be renewed annually between October 1 and December 31, and establishments must pay the annual establishment registration fee. Failure to register can result in import alerts, warning letters, and injunctions.
Key FDA Requirements for Registered Warehouses
• Proper facility design: Well-defined zones for different product types, minimizing cross-contamination risks and separating high-risk from lower-risk items.
• Hygiene standards: Strict cleaning protocols, pest control programs, and continuous environmental monitoring.
• Documented procedures: Written SOPs covering receipt, storage, distribution, and recall processes.
• Personnel training: All staff handling regulated devices must be trained and records maintained.
To learn how a purpose-built facility manages these obligations end-to-end, explore our medical device fulfillment services page.
21 CFR Part 820: Quality System Regulation (QMSR)
The FDA’s Quality System Regulation under 21 CFR Part 820, now updated as the Quality Management System Regulation (QMSR) with a February 2, 2026 effective date, is the primary federal framework governing how medical device warehouses must operate. The QMSR aligns U.S. current Good Manufacturing Practice (cGMP) requirements with ISO 13485:2016, creating a single harmonized quality standard for global operations.
21 CFR 820.150, Storage Requirements Specifically
Section 820.150 of the QSR directly addresses warehouse operations and mandates:
• Control of storage areas and stockrooms to prevent mix-ups and contamination
• Special handling for products susceptible to deterioration over time, including expiry date management
• Systematic procedures for receipt and dispatch authorization
• Regular review of stored inventory to detect deterioration
• Documentation of all storage area activities for traceability and audit purposes
Regulatory Alert: Under QMSR (effective February 2, 2026), 3PLs operating under a device manufacturer’s establishment registration must be audit-ready against the internationally harmonized ISO 13485 standard. If your warehouse partner cannot produce QMS documentation on demand, you have a compliance gap.
Our team of fulfillment specialists helps device brands build compliant warehouse workflows. Discover our full-spectrum order fulfillment solutions designed specifically for regulated industries.
ISO 13485: The Gold Standard for Medical Device QMS
ISO 13485 is an internationally recognized quality management system standard specific to the medical device industry. While ISO 9001 covers general manufacturing quality, ISO 13485 adds device-specific requirements for risk management, regulatory compliance, and post-market surveillance that are critical for warehousing partners.
Why ISO 13485 Matters for Warehousing
• Demonstrates commitment to regulatory compliance beyond the minimum required by law
• Required or strongly preferred by most multinational device manufacturers when qualifying logistics partners
• Harmonized with FDA QMSR under 21 CFR Part 820, simplifying dual-market compliance
• Provides a structured framework for corrective and preventive actions (CAPA), essential during product recalls
• Required for distribution into the EU under the EU Medical Device Regulation (EU MDR)
What ISO 13485 Certification Means for Storage Operations
An ISO 13485-certified warehouse must maintain:
• Documented QMS: A quality manual, procedures, work instructions, and records covering every warehouse function.
• Management review: Regular senior-level review of quality objectives and performance metrics.
• Internal audits: Scheduled self-audits against ISO 13485 requirements, with corrective actions tracked to closure.
• Supplier qualification: Formal evaluation and approval of sub-suppliers including carriers, packaging vendors, and technology providers.
• Complaint handling: Processes for receiving, investigating, and reporting product complaints to the device manufacturer.
When evaluating a 3PL fulfillment partner for medical devices, always request ISO 13485 certification documentation and verify its current scope covers warehousing and distribution activities.
Temperature and Environmental Controls
Temperature excursions are one of the leading causes of medical device quality failures and FDA enforcement actions. From in vitro diagnostics to biological specimens, reagents, sterilants, and implantable devices, precise environmental control is both a patient safety requirement and a regulatory mandate.
FDA Environmental Control Requirements
Under 21 CFR Part 820, warehouses must implement:
• Validated storage environments: Temperature and humidity ranges must be validated and mapped across the storage area.
• Continuous monitoring systems: Automated sensors with alarm capabilities, not manual spot-checks, are required for high-risk products.
• Documented temperature records: Facilities must maintain comprehensive records to demonstrate compliance during audits.
• Emergency procedures: Documented protocols for handling power outages, equipment failures, or natural disasters that could compromise storage conditions.
Temperature Requirements by Device Category
| Device Category | Storage Temperature | Key Compliance Note |
|---|---|---|
| Standard devices (Class I) | Ambient (15–25°C) | Humidity control required |
| In vitro diagnostics & reagents | 2–8°C (refrigerated) | Validated cold chain, continuous monitoring |
| Biological specimens | Below −20°C (frozen) | Dedicated cryogenic storage, alarm systems |
| Sterilants & disinfectants | Manufacturer-specified | Segregated storage, ventilation required |
| Implantable devices (Class III) | Controlled ambient | Full traceability, unit-level serialization |
Choosing a medical device fulfillment services provider with pre-validated, continuously monitored storage environments eliminates the capital investment and regulatory burden of building these systems in-house.
UDI Compliance and Lot Traceability
The Unique Device Identification (UDI) system is the FDA’s cornerstone traceability program for medical devices. Mandated under 21 CFR Parts 801 and 830, UDI requires that every medical device carry a unique identifier in both human-readable and machine-readable (barcode or RFID) formats.
UDI Warehouse Requirements
• Label integrity: UDI labels must be preserved on all inbound and outbound shipments without alteration or damage.
• GUDID database registration: Class II and III devices must be registered in the FDA’s Global Unique Device Identification Database (GUDID), and warehouse records must match GUDID entries.
• Scanning and verification at receipt: Every device must be scanned upon arrival to confirm UDI integrity before entering storage.
• Lot and serial number tracking: Class II and III devices require lot-level and unit-level traceability from receipt through final delivery.
• Expiry date management: FEFO (First Expired, First Out) inventory management is mandatory for products with shelf-life limitations.
Why Traceability Is Non-Negotiable
In the event of a product recall, your warehouse’s ability to produce chain-of-custody records within hours, not days, determines the speed and scope of the recall response. Incomplete traceability translates directly into expanded recalls, regulatory penalties, and reputational damage.
A purpose-built 3PL fulfillment solution for medical devices will have lot tracking, UDI verification, and chain-of-custody documentation built into standard operating procedures, not bolted on after onboarding.
Security, Access Control, and Contamination Prevention
Physical security is a critical and often underestimated, component of medical device warehouse requirements. Unauthorized access, tampering, and contamination are all regulatory concerns as much as safety ones.
Physical Security Requirements
• Restricted access areas with electronic access control logs for all entry and exit events
• 24/7 security monitoring with CCTV coverage of storage and staging areas
• Tamper-evident packaging protocols and procedures for handling suspected tampering
• Segregated quarantine zones for returned, damaged, or non-conforming product
• Visitor access logs and escort procedures for all non-credentialed personnel
Contamination Prevention
• Cleaning and sanitation schedules: Documented programs with verification records for all storage areas.
• Pest control programs: Third-party certified pest management with regular documentation and inspection logs.
• Product segregation: Dedicated zones for different device classes, sterile products, and hazardous materials, with defined airlock or transition protocols.
• Gowning and hygiene requirements: Personnel handling sterile devices must follow documented hygiene procedures.
These standards are integrated into every order fulfillment solution we provide for healthcare and medical device clients.
Documentation, SOPs, and Audit Readiness
In regulated industries, if it isn’t documented, it didn’t happen. A medical device warehouse’s documentation program must be thorough enough to satisfy FDA inspectors, ISO auditors, and the device manufacturer’s own quality team simultaneously.
Essential Documents for Every Medical Device Warehouse
• Quality Manual: Top-level document describing the QMS scope, policy, and framework.
• Standard Operating Procedures (SOPs): Step-by-step instructions for every critical warehouse function, from receiving and storage to pick, pack, ship, and returns.
• Work Instructions: Granular task-level guidance for frontline staff, including visual aids where appropriate.
• Receiving and inspection records: Documentation of every inbound shipment, including condition, quantity, lot number, and UDI verification results.
• Storage monitoring logs: Continuous temperature and humidity records with automated alerts and exception reports.
• Non-conformance reports (NCRs): Formal records of any product, process, or equipment that does not meet specifications.
• CAPA records: Corrective and preventive action documentation tracking root cause analysis through closure.
• Training records: Personnel training completion records aligned to current SOPs.
• Audit reports: Internal and external audit findings with corrective action plans.
Audit Tip: FDA inspectors conducting warehouse audits focus heavily on the consistency between SOPs and actual practice. If your written procedures describe a process that staff perform differently, that gap will appear as a 483 observation. Regular internal audits that compare documented procedures to observed operations are essential.
Packaging and Labeling Requirements
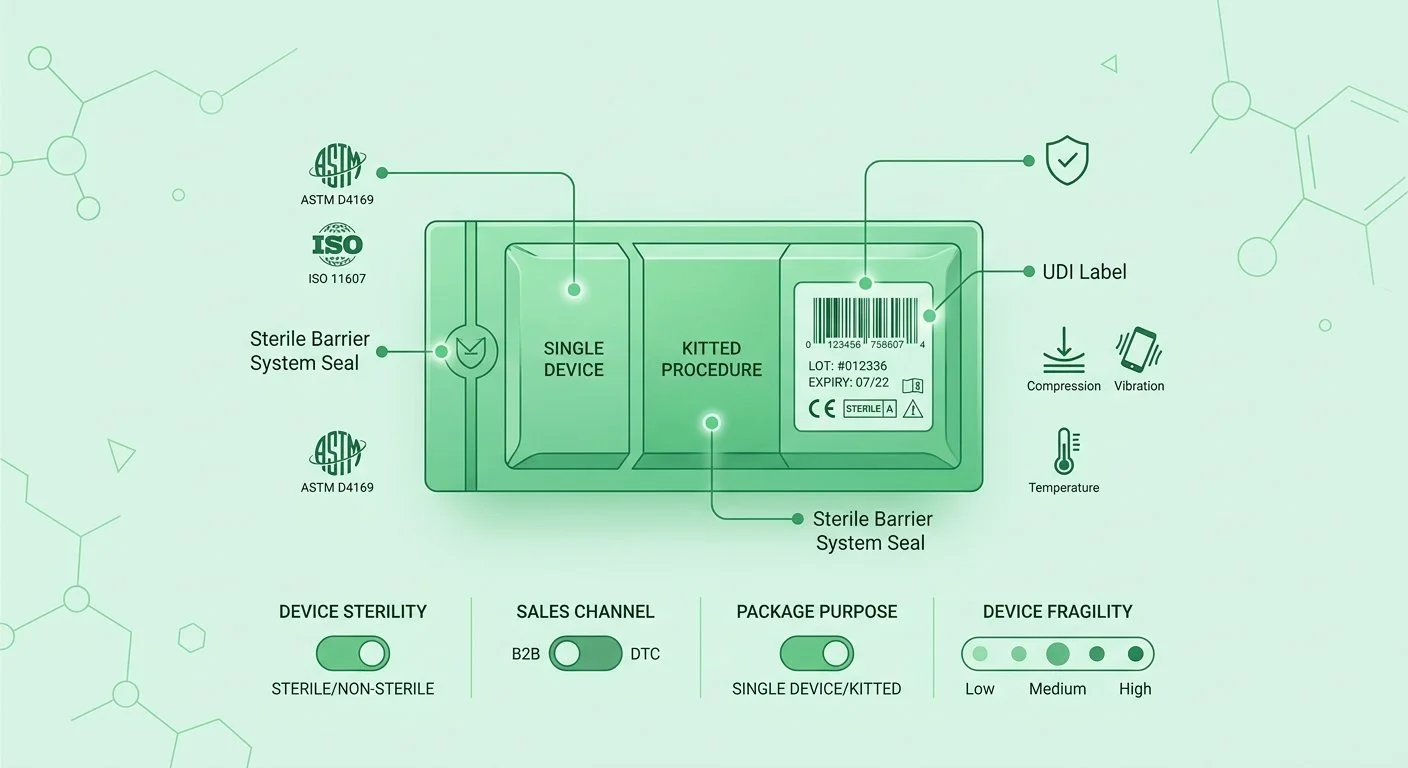
Packaging for medical devices is not a cost-optimization exercise, it is a compliance and liability function. The FDA requires that device packaging maintain sterile barriers, prevent physical damage, and preserve product integrity throughout the distribution chain.
Packaging Requirements
• Sterile barrier system integrity: Packaging must be validated to maintain sterility from manufacture through point of use.
• ASTM and ISO packaging standards: Relevant standards include ASTM D4169 (distribution simulation), ISO 11607 (sterile barrier systems), and ISTA protocols.
• Damage protection: Packaging must protect against physical damage from compression, vibration, and temperature during transit.
• Labeling compliance: All outer and inner packaging must display complete UDI, lot number, expiry date, storage instructions, and regulatory symbols.
Special Packaging Considerations
• Kitting and assembly: Medical device kits require documented bill-of-materials and assembly verification against quality-approved specifications.
• Custom packaging for B2B and DTC channels: Channel-specific packaging must maintain all regulatory requirements while meeting customer presentation standards.
Ourmedical device fulfillment services team includes certified packaging specialists experienced in FDA-compliant sterile barrier packaging, procedure kit assembly, and UDI-compliant labeling for all device classes.
Class I, II, and III Device Storage: What Changes?
FDA device classification directly determines the level of warehouse compliance required. Understanding the differences between Class I, II, and III device storage requirements is essential when evaluating facilities or building out fulfillment workflows.
Class I Devices
Examples: bandages, examination gloves, tongue depressors, handheld surgical instruments.
• Basic storage and chain-of-custody records required
• General controls apply; premarket notification typically not required
• Lower traceability burden: lot-level tracking recommended but unit serialization generally not required
Class II Devices
Examples: powered wheelchairs, infusion pumps, surgical drapes, pregnancy test kits.
• 510(k) clearance required; documentation must be maintained at the warehouse
• UDI compliance and GUDID registration mandatory
• Lot-level traceability required at all transfer points
• Additional special controls may specify labeling, post-market surveillance, or environmental storage requirements
Class III Devices
Examples: implantable cardiac pacemakers, cochlear implants, deep brain stimulators.
• Pre-Market Approval (PMA) records must be accessible at each transfer point
• Unit-level serialization and traceability required
• Strictest environmental controls, with validated and continuously monitored storage
• Full chain-of-custody documentation from manufacturer through implanting facility
Before accepting inventory: Always verify device classification before warehousing any FDA-regulated product. Misclassification or failure to verify can expose your facility to enforcement action even if the device manufacturer made the error.
For expert guidance on handling all three device classes under a singleorder fulfillment solution, our team can help you build compliant workflows that scale with your product portfolio.
Choosing a 3PL for Medical Device Warehousing
Selecting the right 3PL fulfillment partner is one of the highest-stakes decisions a medical device company makes. The wrong partner doesn’t just create operational problems, it can trigger FDA enforcement actions, product recalls, and patient safety events.
Non-Negotiable 3PL Qualifications for Medical Devices
• FDA registration: The facility must be registered with the FDA under the appropriate establishment code for the device classes you distribute.
• ISO 13485 certification (or QMSR compliance): Certification scope must explicitly cover warehousing and distribution, not just manufacturing.
• Demonstrated UDI and lot tracking capabilities: These must exist in standard SOPs, not offered as add-on services after onboarding.
• Temperature-validated storage environments: Validation protocols and monitoring systems appropriate for your device’s storage requirements.
• Documented CAPA and NCR programs: The ability to identify, document, and resolve quality issues in a traceable manner.
• Audit history and inspection readiness: Willingness to share recent FDA inspection records, ISO audit reports, and client quality agreement templates.
Evaluating Technology and Integration Capabilities
• WMS with lot and serial number tracking: Real-time inventory visibility at the lot and unit level, accessible to your quality team.
• ERP and QMS integration: API or EDI connectivity to your existing systems without requiring custom development projects.
• Automated environmental monitoring: Continuous data streams with configurable alert thresholds and automated reporting.
• Recall simulation capability: The ability to identify and isolate all units of a specific lot within hours in a mock recall exercise.
A modern3PL fulfillment approach combines logistics execution with quality management technology, allowing device manufacturers to maintain compliance oversight while scaling without being locked into inflexible infrastructure.
Key Questions to Ask Your Warehouse Partner
When qualifying a potential 3PL or warehouse for medical device storage, ask these questions directly and require documented evidence for the answers:
1. Can you produce your FDA establishment registration number and confirm it covers our device classes?
2. Is your ISO 13485 certification current, and does the scope explicitly include warehousing and distribution?
3. What temperature-validated storage zones do you maintain, and can you share the validation protocols?
4. How do you manage lot tracking and UDI verification at receipt, storage, and dispatch?
5. Can you conduct a mock recall exercise and identify all units of a given lot within 4 hours?
6. What does your CAPA program look like, and can you share a recent example of a corrective action from root cause to closure?
7. Have you received any FDA Form 483 observations or warning letters in the past three years? If so, what were the findings and corrective actions?
8. Can your WMS integrate with our ERP and QMS without requiring a multi-month custom development project?
If a prospective partner cannot answer all of these questions with documented evidence, continue your search. Themedical device fulfillment services team at Rush Order can address each of these areas with direct documentation.
How Rush Order Meets Every Medical Device Warehouse Requirement
Rush Order is built specifically to serve regulated industries, and ourmedical device fulfillment services go beyond standard 3PL capabilities. Here’s how we align with every major medical device warehouse requirement:
Regulatory Compliance
• FDA-registered facility: Our establishments are registered with the FDA and maintained in current good standing.
• 21 CFR Part 820 / QMSR compliant: All warehouse operations are conducted under a documented quality management system aligned with QMSR requirements.
• ISO 13485-aligned QMS: Our quality framework aligns with ISO 13485:2016 across warehousing, distribution, and value-added services.
Traceability and Technology
• Lot and serial number tracking at every transfer point: Automated scanning at receipt, storage, pick, pack, and ship for complete chain-of-custody.
• UDI verification built into receiving SOPs: Every inbound shipment is scanned and verified against GUDID records before entering inventory.
• FEFO inventory management: Automated first-expired-first-out logic to prevent use of expired product.
• ERP and QMS API integration: Seamless connectivity to your existing systems for real-time inventory visibility.
Environmental and Physical Controls
• Temperature-validated storage zones: Ambient, refrigerated, and frozen storage areas with continuous automated monitoring.
• 24/7 environmental alarm systems: Immediate alerts to designated personnel for any temperature or humidity excursion.
• Segregated storage for device classes: Dedicated zones for Class I, II, and III devices with appropriate access controls.
Scalable Order Fulfillment Solutions
Ourorder fulfillment solutions are designed to scale with your growth whether you’re shipping 100 units per month or managing a nationwide distribution network. We support DTC, B2B, hospital direct, and clinic distribution channels from a single compliant inventory pool.
We also offer kitting and assembly services for procedure kits, equipment bundles, and starter sets, with documented BOM verification and quality inspection records for every build. Explore our complete3PL fulfillment solutions to see how we can support your distribution network.
Frequently Asked Questions
What regulations govern medical device warehousing in the United States?
Medical device warehousing in the U.S. is primarily governed by 21 CFR Part 820 (QMSR), FDA registration requirements under 21 CFR Part 807, and UDI regulations under 21 CFR Parts 801 and 830. Additional regulations may apply depending on device type, including IATA Dangerous Goods Regulations for devices with lithium batteries or radioactive components, and HIPAA for devices that store or transmit protected health information.
Does a 3PL warehouse need to be FDA registered?
Yes, if the 3PL operates under a device manufacturer’s establishment registration and performs activities such as repackaging, relabeling, or storage as part of the distribution chain. The level of registration required depends on the specific activities performed and the device classes handled. Always confirm FDA registration status before onboarding a warehouse partner, and review ourmedical device fulfillment services page for details on our current registration status.
What is ISO 13485 and is it required for medical device storage?
ISO 13485 is a quality management system standard specific to the medical device industry. While it is not legally mandated by the FDA, it is functionally required for the EU market and is expected by most multinational device manufacturers when qualifying distribution and warehousing partners. Under QMSR (effective 2026), U.S. requirements are now harmonized with ISO 13485, making certification increasingly important for all 3PLs handling medical devices.
What is FEFO inventory management and why does it matter?
FEFO stands for First Expired, First Out. It is the inventory management methodology required for medical devices and pharmaceuticals with shelf-life limitations. Unlike FIFO (First In, First Out), FEFO prioritizes shipment of products closest to their expiry date regardless of receipt date, preventing the distribution of expired product. Proper FEFO management requires WMS-level automation and is non-negotiable for any FDA-regulated product category.
How do I find a compliant 3PL for medical device fulfillment?
Start by confirming FDA registration, ISO 13485 certification scope, and UDI tracking capabilities. Request documentation of recent audits and FDA inspection history. Then evaluate technology integration, environmental monitoring infrastructure, and CAPA program maturity. Ourorder fulfillment solutions team specializes in regulated industries and can walk you through our compliance documentation in detail.
Conclusion: Build Your Compliance Foundation Before You Ship
Medical device warehouse requirements are not bureaucratic obstacles, they are the infrastructure that protects patients, brands, and supply chains. From FDA registration and QMSR compliance to ISO 13485 certification, UDI traceability, temperature control, and rigorous documentation, every requirement exists because the stakes of failure are measured in patient outcomes.
The good news: you don’t have to build this infrastructure from scratch. A purpose-builtmedical device fulfillment services provider brings pre-validated systems, experienced staff, and documented quality programs that would take years and millions of dollars to replicate internally.
Whether you’re launching a new device, scaling an existing distribution network, or re-evaluating your current warehouse partner after a quality event, our team is ready to help. Explore our full range oforder fulfillment solutions, review our specialized3PL fulfillment services, or go directly to ourmedical device fulfillment services page to see how Rush Order meets every requirement covered in this guide.